
Die offizielle Bezeichnung der Medizinprodukte-Verordnung ist „MDR 2017 | 745“. Diese stellt gewerbliche Labore, Praxislabore und Serienhersteller vor noch nie da gewesene regulatorische Herausforderungen. Es steht fest, dass die Medizinproduktverordnung für alle Hersteller von Medizinprodukten verpflichtend ist. Das schließt gewerblichen Dentallaboren und Praxislaboren jeder Größe ein. Auf eine „Kleine Anfrage“ der FDP-Bundestagsfraktion im Juni 2019 antwortete die Bundesregierung sehr klar und deutlich: „Ähnlich wie für die Hersteller werden mit der MDR auch die Anforderungen an Sonderanfertiger erhöht. […] Besondere Herausforderungen für KMU, die Sonderanfertigungen herstellen, bestehen in diesem Zusammenhang in den Anforderungen der MDR in Bezug auf die klinische Bewertung (inklusive der klinischen Nachbeobachtung), des Risikomanagements sowie der proaktiven Überwachung nach dem Inverkehrbringen […]“
Hintergrund der Verordnung
Die Europäische Kommission hat 2008 über einen neuen Rechtsrahmen für Medizinprodukte diskutiert. Die ersten Entwürfe von jenem Rechtsrahmen wurden 2012 veröffentlicht. Die bis zu diesem Zeitpunkt bestehenden Grenzen der Richtlinien (92/43 EWG und das Medizinproduktegesetz), wiesen diverse Probleme mit Implantaten, wie beispielsweise Metall-Metall Hüftprothesen, auf. Ein mechanisch-korrosiver Abrieb in den Prothesen führte dazu, dass eine erhöhte Konzentration von Kobalt- und Chrom-Ionen im Gewebe nachgewiesen werden konnte. Die betroffenen Produkte wurden vom Hersteller DePuy 2010 zurückgerufen. Vorangetrieben wurde die Änderung des Rechtsrahmens für Medizinprodukte jedoch in erster Linie durch die Bekanntgabe einer der größten Skandale der Medizinproduktbranche, mit etwa 400.000 betroffenen Personen in 65 Ländern: Der sogenannte „PIP-Skandal“ um minderwertige Brustimplantate, welcher weltweit Aufsehen erregt hat.
Die Europäische Kommission reagierte auf den PIP-Skandal, in dem die 2008 angekündigte Revision des Rechtsrahmens für Medizinprodukte weiter vorangetrieben wurde. Am 26. September 2012 wurden die ersten Entwürfe der Verordnung für Medizinprodukte und In-Vitro Diagnostika von der Europäischen Kommission präsentiert. Das vorläufige Ziel war, den Entwurf als Gesetzestext bis Ende 2014 zu übernehmen. Die endgültige Verabschiedung der neuen Verordnung sollte jedoch bis April 2017 andauern. Die dreijährige Übergangsfrist zur Implementierung wurde im April 2020 aufgrund der Corona Pandemie um ein Jahr verlängert. So müssen alle Medizinproduktehersteller die Anforderungen bis zum 26. Mai 2021 umgesetzt haben.
Bei der Verordnung handelt es sich um eine Schutzmaßnahme, die den Verbraucher bzw. Patienten in den Fokus stellt. Dabei werden europaweit alle medizinischen Wirtschaftsakteure und Gesundheitseinrichtungen miteinbezogen, da sie für den gesamten Lebenszyklus eines Medizinprodukts verantwortlich sind. Zusammenfassend heißt dies, dass jeder der an dem Wertschöpfungsprozess beteiligt ist, somit auch für die vor- bzw. nachfolgende Stufe des Prozesses (mit-)verantwortlich ist.
Anforderungen an Hersteller von Sonderanfertigungen
Die MDR unterscheidet nicht zwischen den Herstellern von Serienprodukten oder den von Sonderanfertigungen. Gemäß Art. 10 der MDR gibt es nur „Hersteller“ im engeren Sinne. Dennoch gelten für Sonderanfertigungen bestimmte Besonderheiten und Einschränkungen. Diese werden im Anhang XIII der MDR erläutert, indem die Verfahrensweise einer Sonderanfertigung beschrieben wird. Von zentraler Bedeutung sind somit die Hersteller im Sinne des Artikels 10 der MDR sowie Sonderanfertiger im Anhang XIII der MDR.
Die zukünftig gesetzlich vorgeschriebenen Anforderungen für Dentallabore sind im Konkreten folgende:
- Risikomanagementsystem, welches folgende Aspekte beinhalten sollte:
- Klinische Bewertung (Sekundär Recherche)
- Klinische Nachbeobachtung (Reklamationen und Kulanzen)
- Sicherheitsbericht (Nachbetrachtung der Maßnahmen)
- Chargenrückverfolgung sicherstellen:
- Die Chargenrückverfolgung ist auftragsbezogen und kann durch die einschlägigen Dentalabrechnungssoftwares abgebildet werden.
Die Dokumentationspflichten
Aus Anhang XIII, Abschnitt 1 der MDR ergeben sich die Dokumentationspflichten für eine Sonderanfertigung. Zudem muss wie bisher eine Konformitätserklärung erstellt werden, die folgende Angaben umfasst:
- Name und Anschrift des Herstellers, sowie aller Fertigungsstätten.
- eine Erklärung, dass das Produkt für einen bestimmten Patient oder Anwender bestimmt ist, der durch seinen Namen, ein Akronym oder einen nummerischen Code identifiziert wird.
- Name des Verordnenden und gegebenenfalls Name der betreffenden medizinischen Einrichtung
- die spezifischen Merkmale des Produkts.
- eine Erklärung, dass das betreffende Produkt den grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der MDR entspricht und gegebenenfalls einen Verweis auf die grundlegenden Sicherheits- und Leistungsanforderungen, welche nicht vollständig eingehalten wurden (mit Angabe der Gründe).
Eine Erklärung, die auf Basis der oben genannten Anforderungen erstellt wurde, muss mindestens für einen Zeitraum von 10 Jahre nach dem Inverkehrbringen des Produktes aufbewahrt werden.
Zusätzlich zu der Erklärung, muss eine Dokumentation von der Herstellung der Sonderanfertigungen erstellt werden, um diese für die zuständige Behörde bereitzuhalten. Die Dokumentation muss zu der Fertigungsstätte bzw. den Fertigungsstätten auch Angaben enthalten, aus denen die Auslegung, die vorhergesehene Leistung, sowie die Herstellung des Produktes ersichtlich wird. Durch diese Angaben lässt sich beurteilen, ob das Produkt den Anforderungen der MDR gerecht wird.
Empfehlung
Es ist zu empfehlen, dass Sie eine sorgfältige Dokumentation der Sonderanfertigungen durchführen. Archivieren Sie jegliche Dokumente, die das Produkt anbetreffen. Zudem sollten Sie auch die Angaben aus der Abstimmung mit dem Zahnarzt und dem Patienten archivieren. Hierzu empfiehlt sich eine digitale Dokumentation mittels Dentalprogramm und Dokumentenkamera.
Führen Sie ein System zur Chargenrückverfolgung ein (z. B. über Lieferscheine, Chargen- und Losnummern). Das System kann praxistauglich mit einer Materialwirtschaft abgedeckt werden.
Risikomanagement- bzw. Qualitätsmanagementsystem
Das zu implementierende Risikomanagementsystem (gelegentlich auch als Qualitätsmanagementsystem bezeichnet) muss gemäß der Verordnung folgende Aspekte abdecken:
- Risikomanagement-Plan für jedes Produkt festlegen, dokumentieren (Produktfamilie)
- Risiken identifizieren, analysieren, bewerten, beseitigen und kontrollieren
- Bewertung der Informationen, die Sie durch die Implementierung des Systems in der Fertigungsphase gewonnen haben
- Gefährdungen und deren Häufigkeit
- Abschätzung der verbundenen Risiken, sowie das Gesamtrisiko
- Nutzen-Risiko-Verhältnis
- Risikoakzeptanz
- Die Erkenntnisse nutzen, um Anpassungen im Herstellungsprozess vorzunehmen
- klinische Bewertung
- klinischen Nachbeobachtung nach dem Inverkehrbringen (z.B. über Reklamationen und Kulanzen)
- Implementierung und Management eines Systems zur Überwachung nach dem Inverkehrbringen gemäß Artikel 83 sowie Prüfung und Dokumentation der Erfahrungen nach dem Inverkehrbringen laut Anhang XIII der MDR (Sicherheitsbericht).
Eine spezielle Norm, sowie eine Zertifizierungsverpflichtung schreibt die MDR nicht vor. Jedoch ist jeder zahntechnische Betrieb dazu verpflichtete ein dynamisches und „gelebtes“ System einzuführen, aufrechtzuerhalten und ständig zu verbessern. (Art. 10 Abs. 9 MDR)
Empfehlung
Verfügen Sie bereits über ein zertifiziertes Qualitätsmanagement, beachten Sie, dass dieses nicht automatisch allen Anforderungen der MDR gerecht wird bzw. erfüllt. Eine Überprüfung Ihres Systems auf die Anforderungen der MDR zur klinischen Bewertung und dem Risikomanagement ist daher zwingend erforderlich.
Hinweis
Die Konformität aller Produkte muss vom Hersteller im Rahmen der MDR nachgewiesen werden: Künftig müssen die Hersteller von Medizinprodukten gewährleisten, dass ihre Produkte beim Inverkehrbringen den Anforderungen der MDR genügen. Wenn die Hersteller dies nicht können, besteht ein Verkehrsverbot. Ein Bußgeld droht außerdem, wenn keine oder eine nicht vollständige Konformitätserklärung nach Anhang XIII der MDR beigefügt wird. Diese Erklärung ist bereits jetzt ein Rechtsakt, der an Bedeutung gewinnen wird. Es muss eine Bestätigung durch das Labor erfolgen, dass dieses gemäß des Anhangs I der MDR ein kontinuierlich geführtes Risikomanagement führt und anwendet.
Klinische Bewertung
Zudem müssen auch Sonderanfertigungen einer klinischen Bewertung unterzogen werden und sind ein Bestandteil des Qualitäts- bzw. des Risikomanagementsystems. Der Umfang des klinischen Nachweises muss den Merkmalen des Produktes und deren Zweckbestimmungen angemessen sein.
Folgende Punkte sind, der klinischen Bewertung betreffend, zu berücksichtigen:
- kritische Bewertung der einschlägigen, derzeit verfügbaren wissenschaftlichen Fachliteratur über Sicherheit, Leistung, Auslegungsmerkmale und Zweckbestimmung des Produkts.
- eine kritische Bewertung der Ergebnisse aller verfügbaren klinischen Prüfungen.
- eine Berücksichtigung der gegebenenfalls derzeit anderwärtig verfügbaren Versorgungsoptionen für diesen Zweck.
Klinische Nachbeobachtung
Für die klinische Nachbeobachtung eines bereits in den Verkehr gebrachten Produktes muss ein Plan festgelegt werden. Für das Sammeln und Bewerten der betreffenden Daten müssen unter anderem folgende Methoden und Verfahren angewendet werden:
- Ermittlung zuvor unbekannter Nebenwirkungen (z. B. Veränderungen infolge Wechselwirkungen mit Medikamenten).
- Bewertung der klinischen Daten zu gleichartigen oder ähnlichen Produkten.
- Einholung des Feedbacks von Anwendern (Reklamationen und Kulanzen).
- Durchsicht wissenschaftlicher Fachliteratur und anderer Quellen klinischer Daten.
Um erforderliche Korrekturen durchzuführen, ist der Hersteller dazu aufgefordert, aus den Erfahrungen der klinischen Nachbeobachtung angemessene Vorkehrung zu treffen. Gemäß des Art. 87 Abs. der MDR, muss den zuständigen Behörden bei vorliegender Kenntnis sofort jedes schwerwiegende Vorkommnis und/oder jede Sicherheitskorrekturmaßnahme im Feld gemeldet werden.
Zudem ist ein Bewertungsbericht von den Ergebnissen aus der klinischen Nachbeobachtung nach dem Inverkehrbringen erforderlich. Um erforderliche Korrekturen durchführen zu können, müssen im Voraus angemessene Vorkehrungen getroffen werden.
Sicherheitsbericht
Jeder Hersteller von Zahnersatz, muss für jede Produktgruppe/Kategorie einen regelmäßigen, aktuellen Bericht über die Sicherheit erstellen: Den sogenannten „Sicherheitsbericht“. Produkte der Klasse IIa, den Produkten des Zahnersatzes, muss der Hersteller in dem Sicherheitsbericht bei Bedarf aktualisieren, jedoch mindestens alle zwei Jahre.
Zeitaufwand für das Implementieren
Die Umsetzung des Risikomanagements und der dazugehörigen Aufgaben kann sehr zeitintensiv sein – muss es aber nicht. Verschiedene Verbände raten zu einer Excel-Lösung und bieten Vorlagen an. Die Hauptarbeit der Einführung des Risikomanagements ist jedoch nicht das Erstellen der Vorlagen, sondern das Verstehen der Verordnung an sich, dem Erheben der Daten und besonders die Analyse und Bewertung der jeweiligen Risikofaktoren innerhalb jeder Produktionsstufe. Jede Stufe gilt es einzeln zu betrachten und zu hinterfragen:
- Welche potenziellen Fehler könnten entstehen?
- Welche Folgen resultieren aus diesen Fehlern?
- Was sind mögliche Fehlerursachen?
- Welche Maßnahmen ergreife ich zur Risikominimierung?
- Wie hoch ist die Wahrscheinlichkeit, dass Fehler auftreten?
- Welche Auswirkungen haben Fehler?
- Wie wahrscheinlich ist es, den Fehler zu entdecken?
Betrachtet man alle Aspekte und die Anzahl an Produktionsstufen, dann wird nachvollziehbar, dass mit einem hohen Zeitaufwand zu rechnen ist. Dieser Zeitaufwand kann vermieden werden, denn Anbieter wie der Dental Risikomanager halten bereits eine erhebliche Bandbreite an bewerteten Positionen vor. Somit erfolgt hier lediglich ein Abgleich, ob Sie als Dentallabor zum selben Ergebnis kommen oder ggf. Anpassungen vornehmen müssen. Dadurch kann eine Zeitersparnis von 95% erreicht werden.
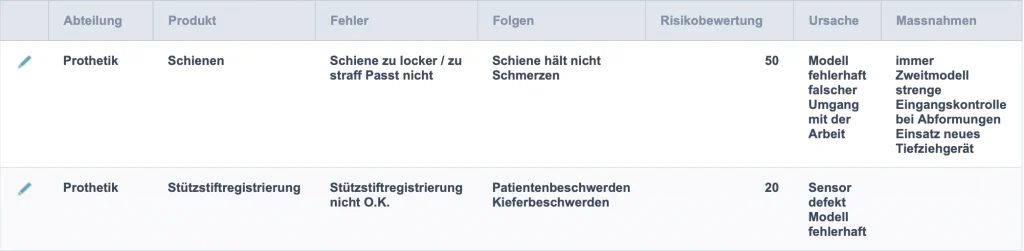
Der aufwendigste Teil der Umsetzung des Risikomanagements ist die Erfassung der Risikopositionen im Dentallabor. Der Dental Risikomanager hat eine Vielzahl dieser bereits erfasst.
Die richtige Software macht den Unterschied
Ratsam ist, sich intensiv mit der Einführung eines Risikomanagementsystems zu befassen. Eine softwaregestützte Umsetzung, wie der Dental Risikomanager, ist mehr als hilfreich und bietet viele Vorteile. Alle Standardarbeitsschritte eines Dentallabors sind bereits im System hinterlegt und müssen nicht zeitintensiv angelegt werden. Das Gleiche gilt für die gängigen Risikoposition. Ferner können Sie ohne Investitionskosten starten und es kann unkompliziert zum jeweiligen Monatsende gekündigt werden.



